What is Cystic Fibrosis?
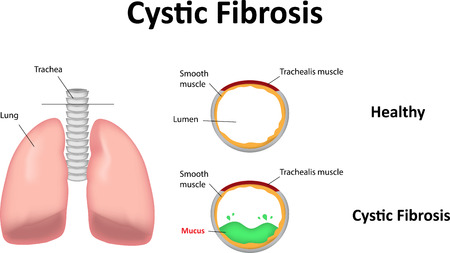
Cystic fibrosis is a genetic disorder. It affects the cells in the lungs that are responsible for the formation of the mucous, sweat and digestive fluids. In normal conditions, the secretions are thin and the main function is to lubricate the organs for normal functioning. In cystic fibrosis, these secretions become excessive and thick (glue like). This causes blockage of the tubules within the lungs. The mucus that collects within the lungs leads to growth of germs and infection within the lungs. This infection leads to cyst formation within the lungs and heals by fibrosis of lungs and scar formation. This exact pathology gave the name to the disease cystic fibrosis (which means cysts and fibrosis in the affected tissues)
Causes of Cystic Fibrosis
Cystic fibrosis causes are mainly genetic in nature. The disorder is caused by genetic mutation in the cftr gene (cystic fibrosis transmembrane conductance regulator). This gene is mainly responsible for regulating the levels of salt and water within the cells and in the intercellular spaces. CFTR works in regulating the digestive juices, mucus and the sweat production. Dysfunction of this gene leads to a disturbance in this function and results in the collection of a sticky substance within the cells which is nothing but thickened up cell secretions. These then become a source of infection in the affected organ.
The pattern of inheritance of cystic fibrosis is autosomal recessive in nature. Both the parents need to have a copy of the mutated gene for it to pass on to the child. If both the parents are carriers then there is a 25% (one in four) chance of the baby getting cystic fibrosis. If only a single copy of the mutated gene is inherited then the child becomes a carrier and does not have the active disease. This child however will pass on the gene to his future generations.
Pathophysiology of Cystic Fibrosis
Cystic fibrosis occurs as a result of mutation in the gene located on the chromosome 7. This particular gene encodes a protein called cftr. When this gene is mutated, the chloride channels within the cells are affected due to which the secretions which are normally thin, become thick and viscous. These thick and sticky secretions collect in the lungs airways and lead to repeated infections. These infections heal by scarring and cause fibrosis of the lungs. Scaring over a large area of the lung tissue leads to breathing troubles.
Defective chloride channels in cystic fibrosis lead to sweat that is extremely salty and leaves stains on the clothes.
When the secretions of the pancreas become thick, it blocks the pancreatic duct. As a result the digestive enzymes do not reach the intestines. This leads to a poor absorption of fat and proteins. The patient suffers from mal absorption and vitamin deficiencies.
Symptoms of Cystic Fibrosis
The symptoms of cystic fibrosis vary from person to person and the intensity depends on the stage at which the disease is. The disease makes its appearance in childhood and infancy. The most common signs and symptoms are:
- Poor growth and weight gain inspite of eating well. This is mainly as a result of malabsorption in the intestines which occurs due to lack of pancreatic enzymes.
- The sweat is excessively salty
- Lung infections that occur frequently.
- Intestinal obstruction in older adults due to blockage of the intestines.
- Thickened secretions in the liver can lead to blockage of the bile duct. Long term liver damage can lead to liver damage and cirrhosis.
- In children there is poor weight gain
- Symptoms like bowel obstruction and meconium ileus in new born babies.
- Vitamin K malabsorption. Normally vitamin K is absorbed from the breast milk and later on from the solid food. In cystic fibrosis, this absorption is poor and so the child has very low stores of vitamin K. Low levels of vitamin K affect the clotting factors in the blood and lead to coagulation disorders. This is a rare condition and it gets diagnosed when the child has unexplained bruising or bleeding that will not stop along with the above mentioned systemic symptoms.
- Scarring and damage to the pancreas as a result of cystic fibrosis can affect the secretion of insulin from the b cells of pancreas. This increases the risk of diabetes in the patients.
- It has been seen that majority of the patients with cystic fibrosis have infertility. This affects both males and females. In males the cause of the infertility is the congenital absence of the vas deference and in females it is the excessive thickening of the cervical mucus or even malnutrition.
- The imbalance in the minerals within the cells may also cause low blood pressure and fatigue in some patients.
Diagnosis of Cystic Fibrosis
(a) Newborn Testing: If there is a history of cystic fibrosis in the parents, the child must be subjected to genetic testing which can confirm the presence of a faulty cftr gene. Blood test may be used to check the functioning of the pancreas.
(b) Sweat Test: Sweat tests are mainly carried out to check the salt content in the sweat of the person suspected to have cystic fibrosis. The doctor will artificially produce sweat on a small patch of the skin on the arms or the legs. The sweat is then collected using a pad and analyzed for the salt content. High salt content indicates cystic fibrosis.
(c) Other Investigations:
- Chest X-ray
- X-ray of paranasal sinuses
- Lung function tests
- Sputum examination and culture
- Prenatal screening of the foetus for cystic fibrosis via amniocentesis
(d) Carrier Testing: If an individual has one normal gene and one faulty gene, then he becomes a carrier of the disease without any active symptoms. There is however a risk of passing on the disease to the future generation. When planning a pregnancy a genetic counselor can help you to check for the risk of transmission of the disease to the fetus by means of a blood or saliva test.

Treatment of Cystic Fibrosis
There is no permanent medical treatment for cystic fibrosis but the main goals of the treatment are to reduce the frequency of lung infections and make the secretions thin so that they do not block the ducts and passages in the various organs. The treatment of cystic fibrosis is multifaceted and so a physician, dietician and physical therapist need to work hand in hand.
- CPT which is also called chest clapping or percussion is performed to loosen the mucous clogging the ducts so that the patient can cough it up. This can be done by using a mechanical precursor or an inflatable therapy vest.
- Aerobic exercise and physiotherapy for cystic fibrosis involves breathing hard so that the mucous logged up in the chest can be coughed up.
- The secretions of the body become very salty so a high salt diet or supplements may be needed to balance the salt levels in the body.
- Medications for cf like anti inflammatory drugs, broncho dilators and mucous thinning medicines are helpful. Antibiotics may be needed to control infections in the lungs.
- Adequate dietary supplements and balanced diet are useful in taking care of the problems caused by malabsorption in intestines of cystic fibrosis patient.
- In severe cases of cystic fibrosis, oxygen therapy may be needed using a mask or prongs.
- Lung transplant may be required if the fibrosis is very wide spread.